Last update: 20251126
Design QV evidence framework
Introduction
Genomic analysis uses many different tools, yet every pipeline must address the same core questions about sequence validity, provenance, normalisation and the evidence supporting a variant. These checks are independent of how the variant was identified or which algorithm produced it.
This guideline defines a shared, tool agnostic specification for reporting that evidence. It provides a minimal set of verifiable data points that any organisation can supply, whether clinical, academic or commercial. Proprietary systems may keep their algorithms private, but they must indicate whether each required evidence check is met. This gives users and regulators a consistent basis for trust while protecting intellectual property.
By separating evidence requirements from implementation, the framework enables interoperability across diverse systems, supports reproducibility and accreditation, and allows the sector to advance without fragmentation.
Conceptual basis
The design builds on the Qualifying Variant (QV) framework by Lawless et al. (2025), which showed that rule definitions can be separated from analysis code and maintained as versioned, portable objects (Figure 1). That approach demonstrated that criteria can be written, cited, audited and reused independently of the internal algorithms that apply them. This guideline generalises the same principle to a downstream set of evidence flags required for Mendelian interpretation, extending the portability and transparency of the QV concept to the full evidence layer. For the framework and suggested file structure see: Lawless, Dylan, et al. “Application of qualifying variants for genomic analysis” medRxiv preprint (2025) | DOI | PDF.
 Figure 1. Shared QV configuration registry. Public or private QV sets define the consensus minimal evidence rules in a portable, versioned format. WGS providers, clinical users, and companies all draw from the same QV database, ensuring that evidence checks are reproducible, transparent, and aligned across independent pipelines.
Figure 1. Shared QV configuration registry. Public or private QV sets define the consensus minimal evidence rules in a portable, versioned format. WGS providers, clinical users, and companies all draw from the same QV database, ensuring that evidence checks are reproducible, transparent, and aligned across independent pipelines.
Two part architecture
The framework is structured around two independent components that work together to provide transparency and reproducibility. The evidence output is generated at the end of secondary analysis and carried forward into tertiary interpretation, allowing end users to verify the essential evidence checks without needing access to the original pipeline.
Part 1: Evidence configuration
A versioned configuration file (YAML/JSON) defines the minimum verifiable evidence set. It specifies the required evidence flags, their definitions, the conditions under which they are valid and the rules for combining them. Because the configuration is external to the pipeline, it can be version controlled, cited, compared and reused across organisations. This separation follows the principle established in the original QV framework.
Part 2: Configuration registry
A registry stores configuration files as stable records with identifiers and checksums. Shared configurations can be used directly, while project specific configurations are preserved for exact reproducibility. The registry may be public or private, as verification depends only on the clarity and stability of the configuration.
Verification model
An analysis system declares the configuration it uses and outputs the completed evidence flags defined in that configuration. Because the configuration is explicit and fixed, users and auditors can verify that the required checks were performed and that the interpretation follows from the stated rules. Internal algorithms remain flexible, but their outputs must conform to the published evidence specification (Figure 2).
Anyone can check:
- which configuration was used
- which evidence flags were produced
- whether the results meet the required checks
This model allows open and closed source pipelines to produce trustable results without disclosing proprietary methods.
 Figure 2. Generation of the evidence flag set at the end of secondary analysis. Each flag is computed directly from reference data and standard checks, independent of the calling algorithm. These flags are then carried forward into tertiary interpretation so that end users can verify the essential evidence without access to the upstream pipeline.
Figure 2. Generation of the evidence flag set at the end of secondary analysis. Each flag is computed directly from reference data and standard checks, independent of the calling algorithm. These flags are then carried forward into tertiary interpretation so that end users can verify the essential evidence without access to the upstream pipeline.
Example: Clinical diagnosis with a private WGS provider
A private company performs WGS and uses a proprietary algorithm to identify a candidate variant in CFTR, an autosomal recessive gene for cystic fibrosis. The internal method is undisclosed, but the clinical team must verify the essential evidence before accepting the result.
Using the framework, the company supplies the minimum evidence state for NM_000492.4:c.1521_1523delCTT (p.Phe508del). A minimal evidence profile may include:
flag_gt_valid: The proband genotype is well formed and passes QC.flag_gt_hom_alt: The proband is homozygous for p.Phe508del, which is compatible with autosomal recessive disease.flag_moi_parent_gt_confirmed_single: One parent is confirmed heterozygous for the same variant, and the other parent is not genotyped. This is sufficient for a homozygous finding in the proband but would be insufficient to confirm a compound heterozygous scenario.flag_moi_consistent_recessive: The combination of a homozygous proband and at least one confirmed carrier parent is consistent with recessive inheritance, even though phase cannot be fully resolved.flag_popfreq_pathogenic_context: The variant is observed in population datasets at frequencies consistent with a known recessive pathogenic allele.flag_known_pathogenic_domain: The deletion affects well characterised functional regions of CFTR with verifiable external evidence from UniProt and related sources.
This set of flags demonstrates that the variant identity is correct, the upstream call is technically valid, the inheritance pattern is sufficiently consistent with autosomal recessive disease, and all required checks have been applied. Crucially, the verification does not depend on how the proprietary algorithm detected the variant.
Any compliant system will produce the same minimal evidence state for this CFTR variant. This ensures that clinical genetics teams receive the information needed to sign off results, while private, academic and clinical providers remain interoperable. Additionally, internal code or intellectual property can remain private (Figure 3).
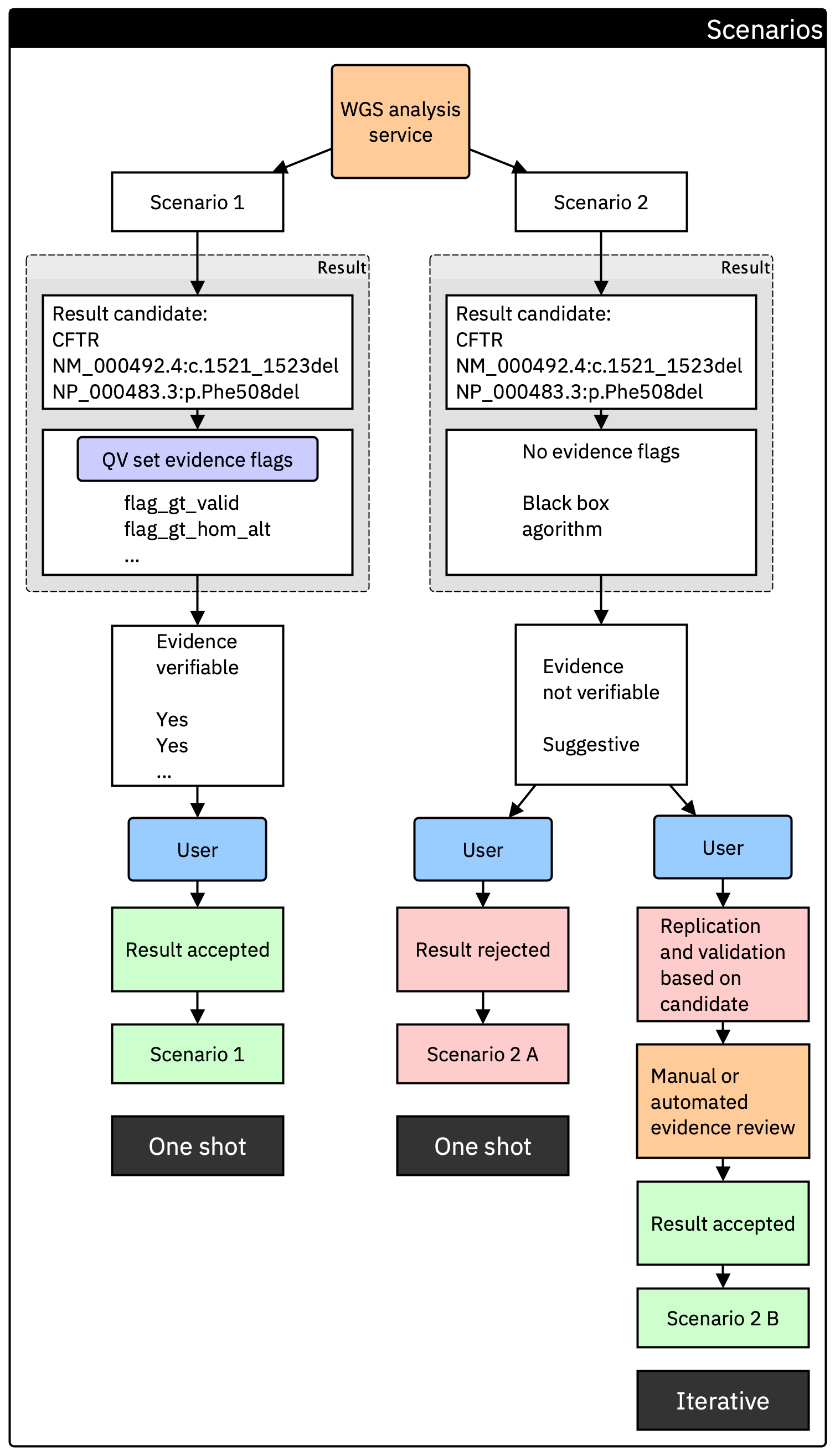
Figure 3. Comparison of two result pathways from a WGS analysis service. In Scenario 1, the provider supplies the minimum evidence flag set, allowing the clinical team to verify the candidate variant in a single step (one shot). In Scenario 2, no evidence flags are provided. The result is either rejected outright or must undergo additional replication and validation, creating an iterative decision process where each variant may need re-evaluation.
Compliance with national and international data standards
The evidence framework is fully aligned with the principles established by SPHN, GA4GH, and national public health digitisation strategies. SPHN requires structured, interoperable, and provenance aware genomic data, and the guideline matches this by defining a computable evidence model, versioned rule sets, and machine readable outputs that can be exchanged across clinical, research, and commercial systems. The use of GA4GH standards, including VRS for variant identity and VA for structured evidence representation, ensures that all evidence states, flags, and provenance fields can be incorporated directly into international data ecosystems without transformation. By adopting these globally recognised models, the framework supports FAIR data practices, federated discovery, and cross institutional interoperability, in line with both Swiss and international objectives for a secure, transparent, and scalable digital health infrastructure.
We generate SPHN compatible RDF metadata in TTL format and store it. SPHN provides the national standard for interoperable health data in Switzerland, so this structure allows direct integration with hospitals, clinics, and national projects. It also aligns with global ecosystems, making connections to systems such as Epic and REDCap straightforward.
RDF expresses data as subject–predicate–object triples, and TTL provides a compact, readable syntax suited for review, versioning, governance, and loading into triple stores.
Features include:
- SPHN compatible classes and relations for variant descriptor, administrative case, and flags like evidence of genotype and mode of inheritance
- TTL exports stored next to the source data for clear audit and governance
- Ready for drop in import into triple stores and analysis pipelines
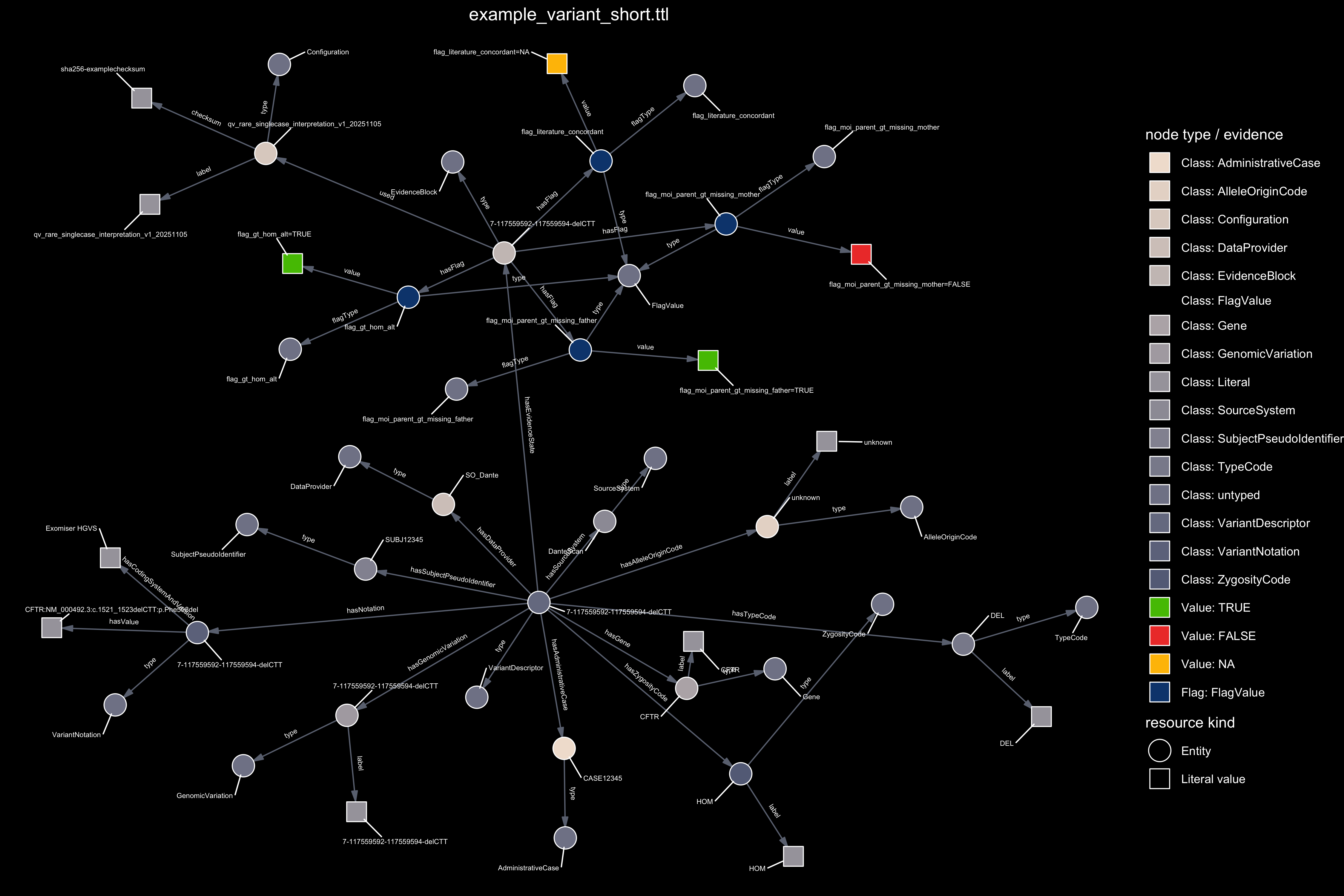
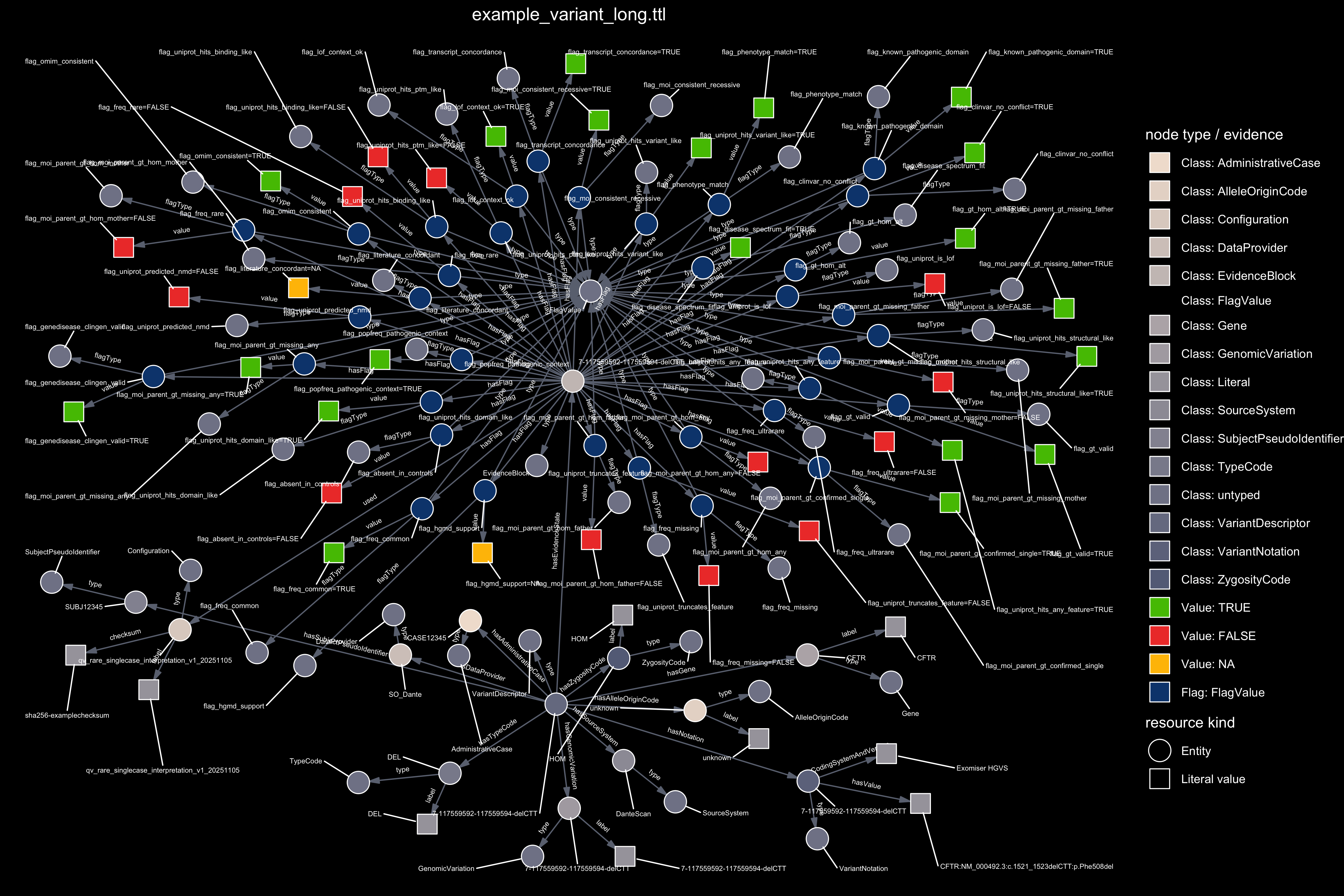
Figure 4. Graph representation of the evidence available for a single variant. The framework applies the reverse logic for clinical genetics: instead of re-questioning whether a variant is pathogenic, we quantify how much verifiable evidence is present to support interpretation. The first panel uses TRUE, FALSE, and NA. to illustrate a minimal example with three evidence flags. The second panel shows the full evidence set produced for the same variant.