Last update: 20240920
Table of contents
Variant features to RDF concept metadata
This process is starting with sequencing_assay, which includes library_preparation, sequencing_run, etc. We will continue through the pipeline until we reach the final end-point required to report a pathogenic variant.
This documentation outlines the transformation of variant information from whole genome sequence (WGS) data to a format adhering to RDF structure data concepts. The aim is to ensure that the omic output from genomic analyses can be seamlessly integrated into clinical data warehouses with high fidelity and clarity.
Number of variables:
- All SPHN RDF concept info (see
SPHN_dataset_release_2024_2_20240502.xlsx) = 1503 - Subset of relevant concepts = 76 (see
example_subset_concepts.tsv) - Relevant WGS pipeline logs = 62 (see
example_report.tsv) Currently automated match = 13 (see
example_report_concepts.Rds)This repository uses a public dataset of example genetic variants and sequencing/analysis log data.
Overview
The process begins with the extraction of variant data from a genomic study, (no sensitive data is included in the public example set). The key variant features such as Chromosome (CHROM), Position (POS), Reference Allele (REF), and Alternate Allele (ALT) are formatted alongside metadata that describes their relationship to RDF concepts. This ensures downstream users can map these data accurately within clinical and research frameworks.
This document is to be updated as we improve the linking of result terms to SPHN_dataset_release_2024_2_20240502.xlsx which is critical so that downstream users can correctly map data.
Aims
- Data preparation: Start with the extracted variant information from the genomic pipeline.
- Key term identification: Focus on essential genomic terms like CHROM, POS, REF, and ALT, Sequencing run, Sequencing instrument.
- Metadata addition: Attach metadata columns that specify RDF concept requirements such as type and cardinality.
- Validation checklist:
- Do we have all necessary variant descriptors present?
- Is there inclusion and accuracy of all metadata explanations?
- Is there alignment of metadata with SPHN omic concepts?
- Downstream users (mapping) can choose from TSV, HTML, JSON, and Rds. Any others needed?
Current version
The observation column is highlighted in GREEN. It contains the data which we report as output from the pipeline for use in our database Here is the completed concept observations (this is file example_report_concepts.html):
| cardinalityViolated | concept_reference_general_concept_name | concept_reference | general_concept_name | observation | release | unique_ID | IRI | active_status_(yes/no) | deprecated_in | replaced_by | concept_or_concept_compositions_or_inherited | general_description | contextualized_concept_name | contextualized_description | parent | type | excluded_type_descendants | standard | value_set_or_subset | meaning_binding | additional_information | cardinality_for_composedOf | cardinality_for_concept_to_Administrative_Case | cardinality_for_concept_to_Data_Provider | cardinality_for_concept_to_Subject_Pseudo_Identifier | cardinality_for_concept_to_Source_System | sensitive_(yes/no) | color_inherited | color_reference | color_observation | color_cardinality |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FALSE | sequencing_assay_sequencing_assay | sequencing_assay | sequencing_assay | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#sequencingassay | yes | NA | NA | concept | an_assay_that_exploits_a_sequencer_as_the_instrument_to_generate_results | sequencing_assay | an_assay_that_exploits_a_sequencer_as_the_instrument_to_generate_results | assay | assay | NA | NA | NA | efo:0003740_|assay_by_sequencer| | NA | NA | 0:n | 1:1 | 0:n | 1:n | NA | #7CCAFF | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_standard_operating_procedure | sequencing_assay | standard_operating_procedure | wgs_with_illumina_novaseq_6000 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasstandardoperatingprocedure | yes | NA | NA | inherited | standard_operating_procedure_associated_to_the_concept | standard_operating_procedure | standard_operating_procedure_that_was_followed_for_this_sequencing_assay | sphnattributeobject | standard_operating_procedure | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #abb1cf | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_predecessor | sequencing_assay | predecessor | kispi_custom_sample_prep_v1 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#haspredecessor | yes | NA | NA | inherited | process_preceding_this_concept | predecessor | sample_processing_preceding_the_sequencing_assay | sphnattributeobject | sample_processing | NA | NA | NA | NA | NA | 0:n | NA | NA | NA | NA | NA | #abb1cf | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_code | sequencing_assay | code | efo_0022396 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hascode | yes | NA | NA | inherited | coded_information_specifying_the_concept | code | code_specifying_the_type_of_sequencing_assay | sphnattributeobject | code | NA | efo;_obi_or_other | for_efo:_descendant_of:_efo:0001455_|assay|;_for_obi:_descendant_of:_obi:0000070_|assay| | NA | NA | 1:1 | NA | NA | NA | NA | NA | #abb1cf | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_identifier | sequencing_assay | identifier | obo:obi_002117_(wgs) | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasidentifier | yes | NA | NA | inherited | unique_identifier_identifying_the_concept | identifier | unique_identifier_identifying_the_sequencing_assay | sphnattributedatatype | string | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #abb1cf | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_start_datetime | sequencing_assay | start_datetime | jul 01 2023 01:01:01 gmt / v0.9.0 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasstartdatetime | yes | NA | NA | inherited | datetime_at_which_the_concept_started | start_datetime | datetime_at_which_the_sequencing_assay_was_first_executed | hasdatetime | temporal | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | yes | #abb1cf | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_data_file | sequencing_assay | data_file | out.fastq | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasdatafile | yes | NA | NA | inherited | data_file_associated_to_the_concept | data_file | data_file_associated_to_the_sequencing_assay | sphnattributeobject | data_file | time_series_data_file | NA | NA | NA | NA | 0:n | NA | NA | NA | NA | NA | #abb1cf | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_sample | sequencing_assay | sample | blood_sample_1 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hassample | yes | NA | NA | inherited | sample_associated_to_the_concept | sample | material_that_is_being_sequenced_by_this_sequencing_assay | sphnattributeobject | sample | tumor_specimen;_isolate | NA | NA | NA | NA | 0:n | NA | NA | NA | NA | NA | #abb1cf | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_library_preparation | sequencing_assay | library_preparation | illumina_truseq_dna_pcr-free | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#haslibrarypreparation | yes | NA | NA | composedof | library_preparation_associated_to_the_concept | library_preparation | the_library_preparation_that_is_part_of_the_sequencing_assay | sphnattributeobject | library_preparation | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_sequencing_instrument | sequencing_assay | sequencing_instrument | a00485 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hassequencinginstrument | yes | NA | NA | composedof | device_associated_to_the_concept | sequencing_instrument | the_device_which_is_used_to_perform_the_sequencing_assay | sphnattributeobject | sequencing_instrument | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_sequencing_run | sequencing_assay | sequencing_run | 334 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hassequencingrun | yes | NA | NA | composedof | sequencing_run_associated_to_the_concept | sequencing_run | sequencing_run_performed_as_part_of_the_sequencing_assay | sphnattributeobject | sequencing_run | NA | NA | NA | NA | NA | 0:n | NA | NA | NA | NA | NA | #92a8d1 | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_intended_read_length | sequencing_assay | intended_read_length | 150 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasintendedreadlength | yes | NA | NA | composedof | intended_read_length_associated_to_the_concept | intended_read_length | the_number_of_nucleotides_intended_to_be_ordered_from_each_side_of_a_nucleic_acid_fragment_obtained_after_the_completion_of_a_sequencing_assay | hasquantity | quantity | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_assay_intended_read_depth | sequencing_assay | intended_read_depth | 30x | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasintendedreaddepth | yes | NA | NA | composedof | intended_read_depth_associated_to_the_concept | intended_read_depth | the_number_of_times_a_particular_locus_(site,_nucleotide,_amplicon,_region)_was_intended_to_be_sequenced_as_part_of_the_sequencing_assay | hasquantity | quantity | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7cac9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_library_preparation | library_preparation | library_preparation | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#librarypreparation | yes | NA | NA | concept | process_which_results_in_the_creation_of_a_library_from_fragments_of_dna | library_preparation | process_which_results_in_the_creation_of_a_library_from_fragments_of_dna | sampleprocessing | sample_processing | NA | NA | NA | obi:0000711_|library_preparation| | NA | NA | 0:n | 1:1 | 0:n | 1:n | NA | #7CCAFF | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_code | library_preparation | code | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hascode | yes | NA | NA | inherited | coded_information_specifying_the_concept | code | code_specifying_the_type_of_library_preparation | sphnattributeobject | code | NA | obi;_efo_or_other | for_obi:_descendant_of:_obi:0000711_|library_preparation| | NA | NA | 0:1 | NA | NA | NA | NA | NA | #abb1cf | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_input | library_preparation | input | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasinput | yes | NA | NA | inherited | input_associated_to_the_concept | input | the_sample_for_which_a_library_is_created | sphnattributeobject | sample | NA | NA | NA | NA | NA | 0:n | NA | NA | NA | NA | NA | #abb1cf | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_output | library_preparation | output | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasoutput | yes | NA | NA | inherited | output_associated_to_the_concept | output | the_ngs_library_that_is_produced | sphnattributeobject | sample | tumor_specimen | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #abb1cf | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_start_datetime | library_preparation | start_datetime | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasstartdatetime | yes | NA | NA | inherited | datetime_at_which_the_concept_started | start_datetime | start_of_library_preparation | hasdatetime | temporal | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | yes | #abb1cf | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_quality_control_metric | library_preparation | quality_control_metric | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasqualitycontrolmetric | yes | NA | NA | inherited | quality_control_metric_associated_to_the_concept | quality_control_metric | quality_control_metric_related_to_the_output_of_the_library_preparation | sphnattributeobject | quality_control_metric | NA | NA | NA | NA | NA | 0:n | NA | NA | NA | NA | NA | #abb1cf | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_predecessor | library_preparation | predecessor | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#haspredecessor | yes | NA | NA | inherited | process_preceding_this_concept | predecessor | process_preceding_this_library_preparation | sphnattributeobject | sample_processing | NA | NA | NA | NA | NA | 0:n | NA | NA | NA | NA | NA | #abb1cf | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_standard_operating_procedure | library_preparation | standard_operating_procedure | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasstandardoperatingprocedure | yes | NA | NA | inherited | standard_operating_procedure_associated_to_the_concept | standard_operating_procedure | standard_operating_procedure_that_was_followed_for_this_library_preparation | sphnattributeobject | standard_operating_procedure | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #abb1cf | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_kit_code | library_preparation | kit_code | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#haskitcode | yes | NA | NA | composedof | coded_information_specifying_the_kit_associated_to_the_concept | library_preparation_kit_code | pre-filled,_ready-to-use_reagent_cartridges_intended_to_improve_chemistry,_cluster_density_and_read_length_as_well_as_improve_quality_(q)_scores_for_this_sample._reagent_components_are_encoded_to_interact_with_the_sequencing_system_to_validate_compatibility_with_user-defined_applications. | hascode | code | NA | efo,_genepio,_fairgenomes_or_other | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_target_enrichment_kit_code | library_preparation | target_enrichment_kit_code | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hastargetenrichmentkitcode | yes | NA | NA | composedof | coded_information_specifying_the_target_enrichment_kit_associated_to_the_concept | target_enrichment_kit_code | indicates_which_target_enrichment_kit_was_used_to_prepare_this_sample._target_enrichment_is_a_pre-sequencing_dna_preparation_step_where_dna_sequences_are_either_directly_amplified_(amplicon_or_multiplex_pcr-based)_or_captured_(hybrid_capture-based)_in_order_to_only_focus_on_specific_regions_of_a_genome_or_dna_sample. | hascode | code | NA | efo,_genepio,_fairgenomes_or_other | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_intended_insert_size | library_preparation | intended_insert_size | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasintendedinsertsize | yes | NA | NA | composedof | intended_insert_size_associated_to_the_concept | intended_insert_size | in_paired-end_sequencing,_the_dna_between_the_adapter_sequences_is_the_insert._the_length_of_this_sequence_is_known_as_the_insert_size,_not_to_be_confused_with_the_inner_distance_between_reads._so,_fragment_length_equals_read_adapter_length_(2x)_plus_insert_size,_and_insert_size_equals_read_length_(2x)_plus_inner_distance. | hasquantity | quantity | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | library_preparation_gene_panel | library_preparation | gene_panel | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasgenepanel | yes | NA | NA | composedof | gene_panel_associated_to_the_concept | gene_panel | collection_of_genes_that_are_the_focus_of_sequencing | sphnattributeobject | gene_panel | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7f6c9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_instrument_sequencing_instrument | sequencing_instrument | sequencing_instrument | a00485 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#sequencinginstrument | yes | NA | NA | concept | a_sequencing_instrument_that_is_used_in_a_sequencing_assay | sequencing_instrument | a_sequencing_instrument_that_is_used_in_a_sequencing_assay | sphnconcept | NA | NA | NA | NA | efo:0000548_|instrument| | NA | NA | NA | 1:1 | NA | NA | NA | #7CCAFF | #f7e7c9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_instrument_code | sequencing_instrument | code | a00485 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hascode | yes | NA | NA | composedof | coded_information_specifying_the_concept | code | code_specifying_the_type_of_sequencing_instrument | sphnattributeobject | code | NA | obi;_efo_or_other | for_obi:_descendant_of:_obi:0400103_|dna_sequencer|;_for_efo:_descendant_of:_efo:0003739_|sequencer| | NA | NA | 1:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7e7c9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_sequencing_run | sequencing_run | sequencing_run | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#sequencingrun | yes | NA | NA | concept | the_valid_and_completed_operation_of_a_high-throughput_sequencing_instrument_associated_with_a_sequencing_assay | sequencing_run | the_valid_and_completed_operation_of_a_high-throughput_sequencing_instrument_associated_with_a_sequencing_assay | sphnconcept | NA | NA | NA | NA | ncit:c148088_|sequencing_run| | NA | NA | NA | 1:1 | NA | NA | NA | #7CCAFF | #f7dac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_identifier | sequencing_run | identifier | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasidentifier | yes | NA | NA | composedof | unique_identifier_identifying_the_concept | identifier | unique_identifier_identifying_the_sequencing_run | sphnattributedatatype | string | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7dac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_datetime | sequencing_run | datetime | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasdatetime | yes | NA | NA | composedof | datetime_of_the_concept | datetime | datetime_the_sequencing_run_was_performed | sphnattributedatatype | temporal | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | yes | #92a8d1 | #f7dac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_read_count | sequencing_run | read_count | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasreadcount | yes | NA | NA | composedof | ready_count_associated_with_to_concept | read_count | the_number_of_sequencing_reaction_results_that_were_pooled_to_assemble_a_sequence_for_a_genomic_region_of_interest_in_a_sequencing_run | hasquantity | quantity | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7dac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_average_insert_size | sequencing_run | average_insert_size | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasaverageinsertsize | yes | NA | NA | composedof | average_insert_size_associated_to_the_concept | average_insert_size | the_average_insert_size_found_during_the_nucleic_acid_sequencing_run | hasquantity | quantity | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7dac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_average_read_length | sequencing_run | average_read_length | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasaveragereadlength | yes | NA | NA | composedof | average_read_length_associated_to_the_concept | average_read_length | the_average_length_for_nucleic_acid_sequencing_reads_generated_in_a_sequencing_run | hasquantity | quantity | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7dac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_mean_read_depth | sequencing_run | mean_read_depth | NA | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasmeanreaddepth | yes | NA | NA | composedof | mean_read_depth_associated_to_the_concept | mean_read_depth | the_number_of_times_a_particular_locus_(site,_nucleotide,_amplicon,_region)_was_sequenced_in_a_sequencing_run | hasquantity | quantity | NA | NA | NA | NA | NA | 0:1 | NA | NA | NA | NA | NA | #92a8d1 | #f7dac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_data_file | sequencing_run | data_file | ../out/example_wgs_sequencing_report_deliverable_summary.tsv | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasdatafile | yes | NA | NA | composedof | data_file_associated_to_the_concept | data_file | data_file_associated_to_the_sequencing_run | sphnattributeobject | data_file | time_series_data_file | NA | NA | NA | NA | 1:n | NA | NA | NA | NA | NA | #92a8d1 | #f7dac9 | #8ed3a0 | #8ed3a0 |
| FALSE | sequencing_run_quality_control_metric | sequencing_run | quality_control_metric | 5f4dcc3b5aa765d61d8327deb882cf99 | 2024.1 | NA | https://www.biomedit.ch/rdf/sphn-schema/sphn/2024/1#hasqualitycontrolmetric | yes | NA | NA | composedof | quality_control_metric_associated_to_the_concept | quality_control_metric | quality_control_metric_associated_with_the_sequencing_run | sphnattributeobject | quality_control_metric | NA | NA | NA | NA | NA | 1:n | NA | NA | NA | NA | NA | #92a8d1 | #f7dac9 | #8ed3a0 | #8ed3a0 |
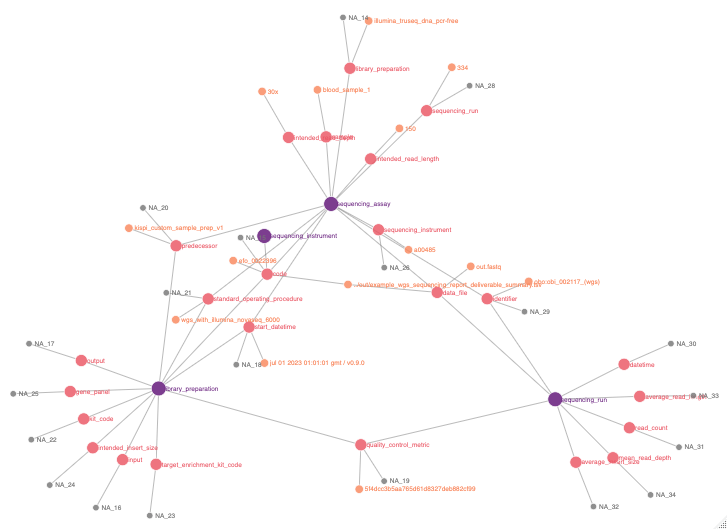
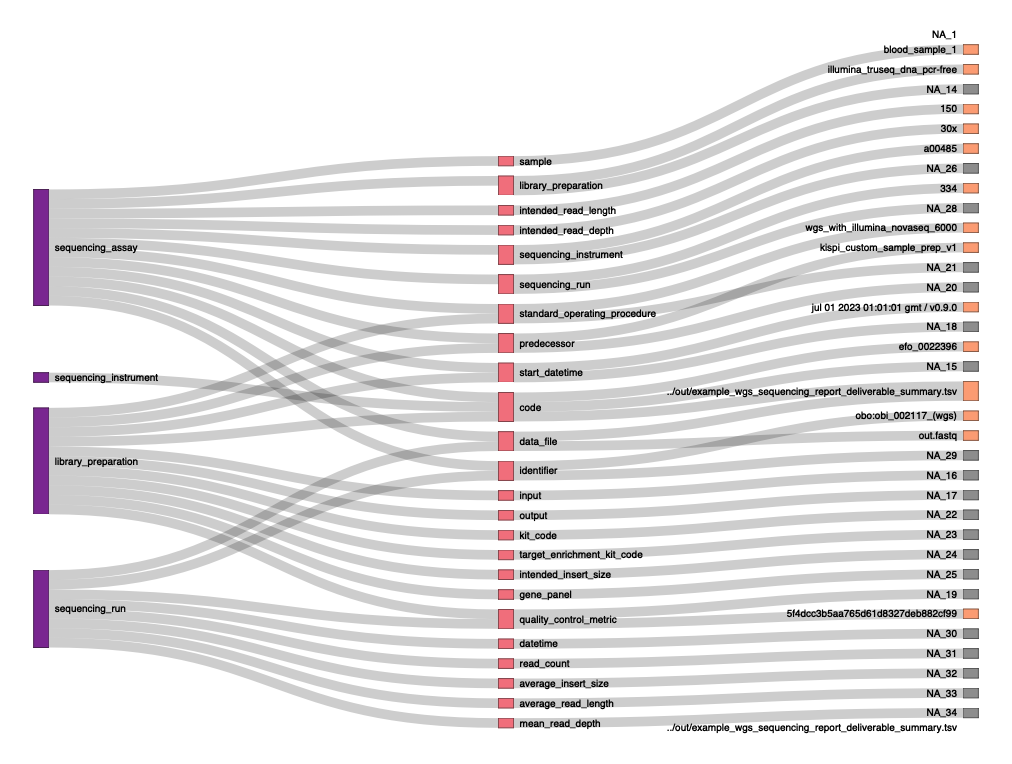
Semantic evidence network
I have added the following method to automatically plot semantic evidence networks which show how evidence provenance has been generated. The dataset is organised into three hierarchical grouping levels based on the column concept_or_concept_compositions_or_inherited. The top level, Level 1, includes entries where this column equals “concept”. The subsequent levels, Level 2 and Level 3, contain entries where this column does not equal “concept”. The distinction between Levels 2 and 3 lies in the presence of distinct observations; Level 3 specifically represents the final observation associated with the general concept names from Level 2, differentiated further by non-empty values in the observation column, making Level 3 essentially a detailed continuation of Level 2.
Nodes within the network are structured with the following attributes: general_concept_name, id, group, name, and observation, where general_concept_name recurs in both Level 2 and Level 3 but differs based on the associated observation. Edges within this hierarchical setup link nodes from Level 1 to Level 2 and from Level 2 to Level 3 using general_concept_name as a consistent link identifier, facilitating a connection between the initial abstract concept level and its more detailed observational breakdowns.
Downloads
Example output (in mutiple filetypes) can be downloaded from the public set:
| File Name | Download Link |
|---|---|
example_report_concepts.tsv | Download |
example_report_concepts.html | Download |
example_report_concepts.Rds | Download |
example_report_concepts.Rds | Download |
example_report_concepts.Rds | Download |
pdf plot_semantic_evidence_plot_network.pdf | Download |
pdf plot_semantic_evidence_plot_sankey.pdf | Download |
html plot_semantic_evidence_plot_network.html | Download |
html plot_semantic_evidence_plot_sankey.html | Download |
Example inputs can be downloaded from the public set:
| File Name | Download Link |
|---|---|
Canton_001_NGS000012345_NA_S46_L001_R1_001.fastq_head.text | Download |
Canton_001_NGS000012345_NA_S46_L001_R1_001_sample_seq_assay_log.text | Download |
SPHN_dataset_release_2024_2_20240502.xlsx | Download |
sequencing assay_van_der_Horst2023.txt | Download |
bwa_10351_101_10453.out.text | Download |
example_variant.Rds | Download |
example_variant.tsv | Download |
Process Steps for Variant Features to RDF Concept Mapping
This section outlines the sequential processing steps from data extraction through to the final merged dataset, prepared for RDF concept mapping. Each step corresponds to a specific script and handles distinct data types or stages in data preparation and merging.
- Export Variant Data from Study
- Extracts variant data from genomic projects focusing on specific genes and filtering for high-impact variants, saving them in formats like RDS and TSV for further processing.
- Read Variant Report Data
- Loads and transforms variant data into a long format to facilitate metadata annotation, preparing the data by adding a column for metadata requirements.
- Read Sequencing Assay Data
- Extracts key sequencing assay data such as identifiers, read depth, and file formats from logs or metadata files, providing crucial context for sequencing parameters.
- Read BWA Read Group Data
- Parses BWA and samtools log files to extract detailed read group information, including metadata about the sequencing run such as machine, file paths, and read group specifications.
- Read Fastq Header Data - Analyzes headers from FASTQ files to extract sequencing instrument details and run metrics, offering a granular look at the sequencing runs which is instrumental in validating sequencing quality and parameters.
- Merge Datasets
- Combines all processed data from the previous steps into a single dataset, aligning them by common identifiers and ensuring consistency across data types.
- Map Pipeline Output to SPHN Concepts
- Maps the merged dataset to standard SPHN RDF concepts, ensuring each data point is correctly classified according to standardized ontology, thus aligning detailed genomic data with broader healthcare data standards.
Terms used in WGS logging
Descriptions for sequencing assay (WGS) terms
| Column Name | Description |
|---|---|
seq_assay_identifier | The unique identifier for the sequencing assay, typically a standard ontology term such as obo:OBI_002117 for Whole Genome Sequencing. |
seq_assay_intended_read_depth | The targeted depth of coverage for the sequencing assay, indicating how many times each base is expected to be sequenced; in this case, 150x. |
seq_assay_intended_read_length | The expected length of each read in the sequencing process, measured in base pairs; here, 20 bp. |
data_file_identifier | Identifier for the data file output from the sequencing, used to trace and access the file within data systems. |
data_file_format | The format of the sequencing data files, specifying the standard used; here, EDAM format 1931, which is typical for FASTQ files from Illumina platforms. |
quality_control_name | The name of the metric used to assess the quality of the sequencing data; in this case, the Phred quality score. |
quality_control_value | The actual quality score achieved, indicating the reliability of the sequencing reads; 78.33% in this context. |
library_prep_kit | Specifies the kit used for preparing DNA libraries for sequencing, critical for understanding the sample preparation methodology; Illumina TruSeq DNA PCR-Free is noted for high fidelity. |
sample_identifier | The unique identifier for the sample being sequenced, used for tracking and reference throughout the sequencing process. |
sample_material_type | The type of biological material from which the sample was derived, with its specific ontology code; snomed:119297000 denotes a blood sample. |
seq_instrument_code | The identifier for the sequencing instrument used, linking to specific equipment details; obo: OBI_0002630 refers to the Illumina NovaSeq 6000. |
sop_name | The name of the Standard Operating Procedure followed during the sequencing, ensuring consistency and reproducibility; here, “WGS with Illumina NovaSeq 6000”. |
sop_description | A brief description of the SOP, providing context and specifics about the sequencing approach used. |
sop_version | The version number of the SOP, which helps in identifying any changes or updates that might affect the sequencing output or interpretation. |
Descriptions for FASTQ/BAM readgroup terms
| Column Name | Description |
|---|---|
START AT | The start timestamp of the sequencing or analysis process. |
END AT | The end timestamp of the sequencing or analysis process. |
sample_read_id | A unique identifier for each sample read in the process. |
rel_dir | Relative directory path where sequencing data is stored. |
dir_id | Directory identifier combining the project and sample ID. |
FILE1 | Path to the first FASTQ file generated by sequencing. |
FILE2 | Path to the second FASTQ file generated by sequencing. |
output_file | Path to the final BAM file generated after processing. |
ID | Internal identifier used to track the sample in analysis. |
SM | Sample name or identifier used within the BAM file. |
PL | Sequencing platform used, indicating technology type. |
PU | Platform unit (PU) tag, often a barcode identifier. |
LB | Library ID which is crucial for distinguishing between libraries prepared differently. |
RG | Read group identifier in a BAM file, encapsulating all other identifiers. |
Descriptions for genetic variants terms
| Column Name | Description |
|---|---|
sample.id | Unique identifier for each sample. |
rownames | Row names corresponding to data entries. |
CHROM | Chromosome number where the variant is located. |
REF | Reference allele at the variant locus. |
ALT | Alternate allele at the variant locus. |
POS | Position of the variant on the chromosome. |
start | Start position of the variant. |
end | End position of the variant. |
width | Width of the variant region. |
Gene | Gene name associated with the variant. |
SYMBOL | Gene symbol. |
HGNC_ID | HUGO Gene Nomenclature Committee ID. |
HGVSp | Human Genome Variation Society protein nomenclature. |
HGVSc | Human Genome Variation Society coding DNA sequence nomenclature. |
Consequence | Consequence of the variant. |
IMPACT | Impact of the variant on the gene or protein function. |
genotype | Genotype showing the variant alleles. |
Feature_type | Type of genomic feature (e.g., transcript, regulatory). |
Feature | Specific feature affected by the variant (e.g., exon, intron). |
BIOTYPE | Biological type of the feature affected (e.g., protein_coding, miRNA). |
VARIANT_CLASS | Classification of the variant based on its genomic context. |
CANONICAL | Indicates if the transcript is the canonical transcript. |
CHROM_Metadata | Type: Chromosome; Cardinality: 1:1; Value Set: SNOMED CT: 91272006, LOINC:48000-4 |
POS_Metadata | Type: Genomic Position; Cardinality: 1:1; Value Set: GENO:0000902 |
REF_Metadata | Type: Reference Allele; Cardinality: 1:1; Value Set: string |
ALT_Metadata | Type: Alternate Allele; Cardinality: 1:1; Value Set: string |