Last update: 20241217
Design DNA SNV INDEL v1
Germline short variant discovery (SNVs + Indels) and interpretation
Protocol name: design_dna_snvindel_v1
Aims
| Phase | Aim | Status | Task |
|---|---|---|---|
| Phase 2 | (1) | Complete | Process all WGS from the study cohort https://www.swisspedhealth.ch to a consensus format. |
| Phase 2 | (2) | 2 of 2 complete | Prepare qualifying variant (QV) sets for each downstream aim. |
| Phase 2 | (3) | v1 complete v2 in progress | Clinical genetics report per individual (i.e., baseline benchmark of known disease-causing). |
| Phase 2 | (4) | 2 experiment complete | GWAS: Statistical genomics to find new cohort-level associations with disease. |
| Phase 2 | (5) | 2 experiment complete | Gene-VSAT: Statistical genomics to find new cohort-level associations with disease. |
| Phase 2 | (6) | 1 experiment complete | Proteome-VSAT: Statistical genomics to find new cohort-level associations with disease (Proteom-VSAT). |
| Phase 2 | (7) | 1 experiment complete | ACAT: Statistical multiomics to find new cohort-level associations with disease.. |
| Phase 2 | (8) | In progress | New methods (ML/DL, causal inference) for individual and cohort-level discovery. |
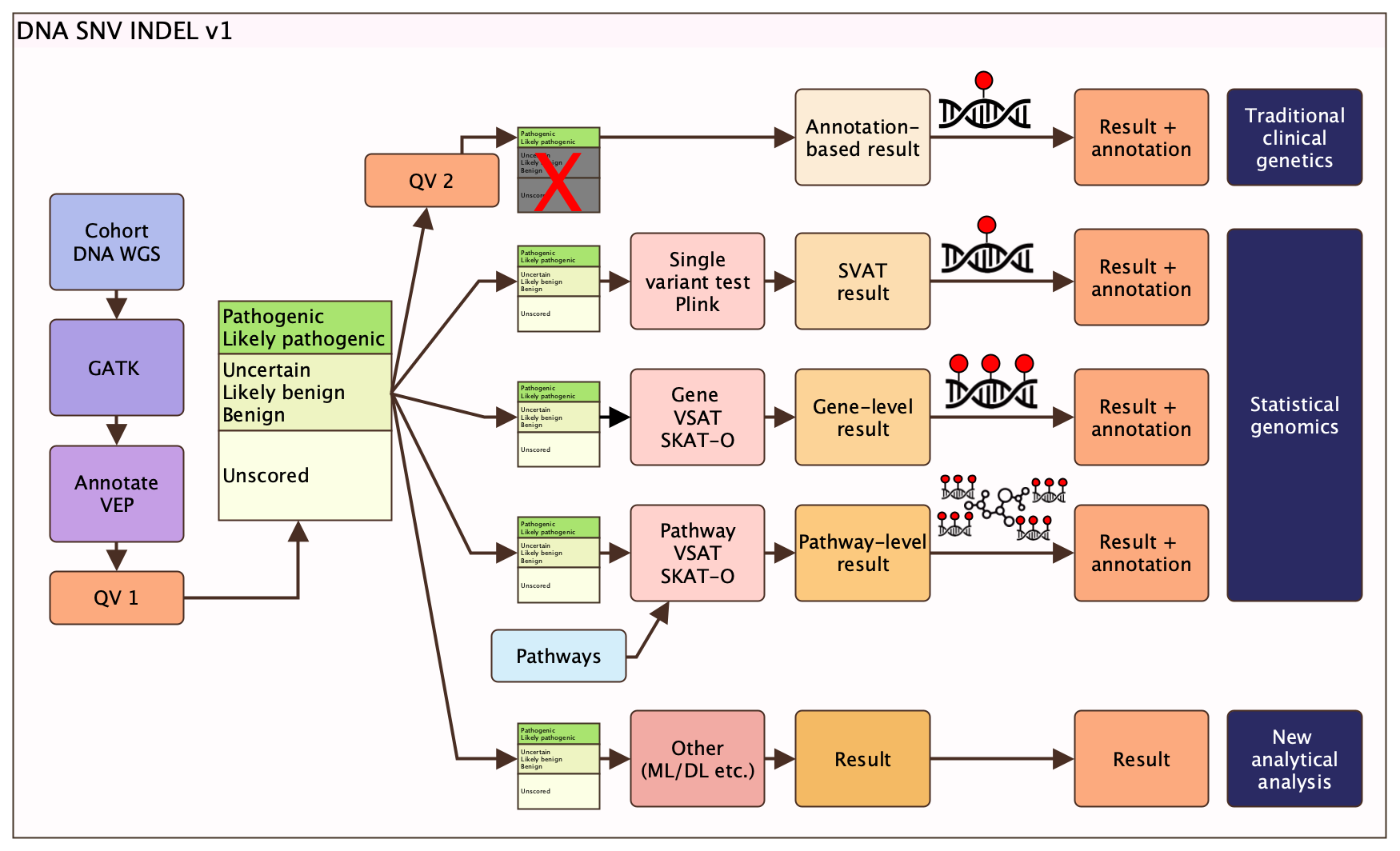
Figure 1: Summary of design DNA SNV INDEL v1 pipeline plan.
Introduction
This protocol is designed to process DNA WGS data in FASTQ format into qualifying qariants (QV) based on consensus variables and thresholds (figure 1). The QV can then be used in multiple applications such as ML/DL to find disease-related variants or gene functions. Additionally, in the clinical genetic protocol further standardised filtering criteria are used to reach a single genetic determinant in a clinical genetics report for each subject. The design name Design DNA SNV INDEL v1 indicates that this protocol is tailored to single nucleotide variants (SNVs) and short insertion/deletions (INDELs) (e.g. GATK pipeline). We implement the genome analysis tool kit GATK best practices workflow for germline short variant discovery (open source licence here). This GATK workflow is designed to operate on a set of samples constituting a study cohort; specifically, a set of per-sample BAM files that have been pre-processed as described in the GATK Best Practices for data pre-processing. Single-variant and genomics-only analysis will be followed up to confirm if causal effects are identified in RNA and protein layers. Joint-multiomic analysis will include all layers in a single statistical model.
Protocol summary
- Process all raw WGS into an analysis-ready format - geonmic VCF (gVCF).
- The first goal is to process all raw whole genome sequencing (WGS) data into analysis-ready formats, specifically into joint cohort Variant Call Format (VCF) using the emit-ref-confidence (ERC) gVCF mode. This involves using a reference model to emit data with condensed non-variant blocks, adhering to the gVCF format. gVCF is split per chromosome.
- The joint cohort chromosome level gVCF are filtered into qualifying variants (QV).
- The QV sets are used individually or mixed to produce the main analysis results:
- QV set 1 for clinical genetics (known disease-causing) report for each individual
- QV set 2 for statistical genomics (new associations with established methods) for cohort level discovery
- QV set 1 or 2 for other methods (ML/DL, causal inference) (new methods) for individual and cohort level discovery
- Release data.
- If not already included in an analysis model, the candidate causal variants will be followed up to confirm if causal effects are identified in RNA and protein layers.
Protocol steps
The major processing steps in sequential order are:
- Aim 1
- FASTQ QC - see DNA QC
- FASTP: QC, check adapters, trimming, filtering, splitting/merging.
- Genome alignment with BWA with reference genome GCA_000001405.15_GRCh38_no_alt_analysis_set
- GATK Duplicates
- GATK BQSR
- GATK Haplotype caller
- GATK Genomic db import
- GATK Genotyping gVCFs
- GATK VQSR
- GATK Genotype refine
- VCF QC - see DNA QC
- Aim 2
- Design QV SNV INDEL V1 Qualifying variants (variables and thresholds) SNV INDEL v1
- Pre-annotation processing: data conversion for simpler handling
- Pre-annotation MAF: filtering to remove noise
- DNA annotation: annotate known effects, biological function, associations
- Aim 3 use a selective mixture of the remaining methods depending on the QV set
- DNA interpretation
- ACMG criteria: Standardised scoring for interpreting variant pathogenicity
- Clinical genetics reports - not documented here
- Aim 4-7 use a selective mixture of the remaining methods depending on the QV set
- Design Statistical genomics v1
- ML/DL projects - not documented here
- Release
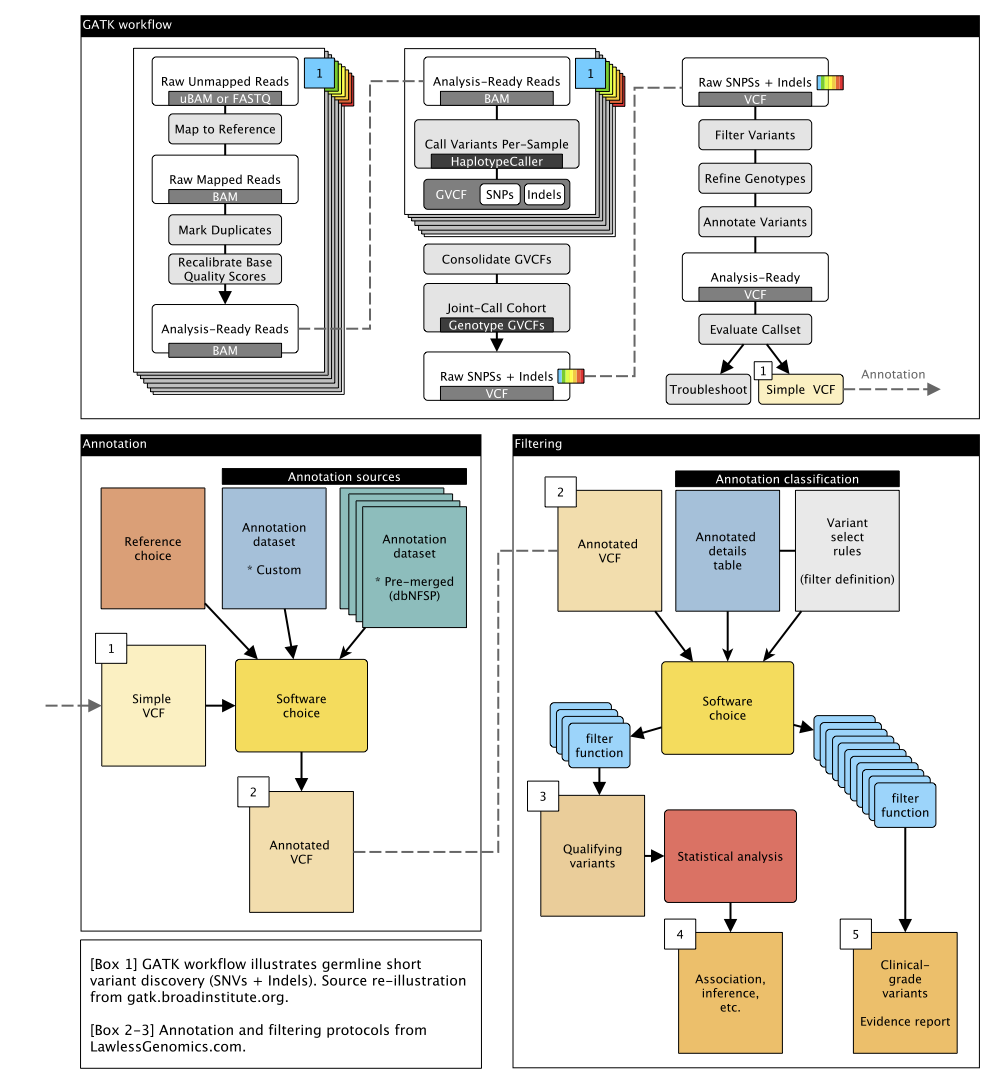
Figure 2: Extended methods of figure 1 DNA germline short variant discovery pipeline plan.
Metrics
Study book data:
CollectWgsMetrics:03b_collectwgsmetrics.sh->study_book/qc_summary_statsmapping, depth, and more. See metrics_collectwgsmetrics.bcftools statsandplot-vcfstats:07c_qc_summary_stats.sh->study_book/qc_summary_statsgVCF summary after HC. See metrics_bcftoolsstats.
Data release
The private internal data release: design_dna_snvindel_v1_release